Înlocuirea liganzilor. Conexiuni complexe ale elementelor d
Una dintre cele mai importante etape în cataliza complexului metalic - interacțiunea substratului Y cu complexul - are loc prin trei mecanisme:
a) Înlocuirea ligandului cu un solvent. Această etapă este de obicei descrisă ca disocierea complexului
Esența procesului în cele mai multe cazuri este înlocuirea ligandului cu un solvent S, care este apoi înlocuit cu ușurință de o moleculă substrat Y.
b) Atașarea unui nou ligand la o coordonată liberă cu formarea unui asociat urmată de disocierea ligandului înlocuit
c) Substituţie sincronă (tip S N 2) fără formare intermediară
În cazul complecșilor Pt(II), viteza de reacție este foarte des descrisă de ecuația cu două căi
Unde k SȘi k Y sunt constantele de viteză ale proceselor care au loc în reacțiile (5) (cu un solvent) și (6) cu ligand Y. De exemplu,


Ultima etapă a celei de-a doua rute este suma a trei etape elementare rapide – eliminarea Cl –, adăugarea de Y și eliminarea moleculei de H 2 O.
În complexele pătrate plate de metale de tranziție, se observă un efect trans, formulat de I.I Chernyaev - influența LT asupra ratei de substituție a unui ligand care se află în poziție trans față de ligandul LT. Pentru complecșii Pt(II), efectul trans crește în seria de liganzi:
H2O~NH3 Prezența efectului trans cinetic și a influenței trans termodinamice explică posibilitatea sintetizării complexelor izomerice inerte de Pt(NH 3) 2 Cl 2: Reacții de substituție electrofilă (SE) a hidrogenului cu un metal în sfera de coordonare a metalului și procesele lor inverse SH – H 2 O, ROH, RNH 2, RSH, ArH, RCCH. Chiar și moleculele de H2 și CH4 participă la reacții de acest tip Reacții de introducere a lui L de-a lungul conexiunii M-X În cazul X=R (complex organometalic), de-a lungul legăturii M-R se introduc și molecule coordonate cu metal (L–CO,RNC,C2H2,C2H4,N2,CO2,O2 etc.). Reacția de inserție este rezultatul unui atac intramolecular al unui nucleofil asupra unei molecule coordonate - sau . Reacții inverse – - și -reacții de eliminare Reacții de adiție oxidativă și de eliminare reductivă M 2 (C 2 H 2) M 2 4+ (C 2 H 2) 4– Aparent, în aceste reacții există întotdeauna o coordonare preliminară a moleculei adăugate, dar aceasta nu poate fi întotdeauna detectată. Prin urmare, prezența unui loc liber în sfera de coordonare sau a unui loc asociat cu un solvent care este ușor înlocuit de un substrat este un factor important care afectează reactivitatea complecșilor metalici. De exemplu, complecșii bis--alil de Ni sunt buni precursori ai speciilor active catalitic, deoarece datorită eliminării reductive ușoare a bis-alilului, apare un complex cu solventul, așa-numitul. nichel „gol”. Rolul locurilor goale este ilustrat de următorul exemplu: Reacții de adiție nucleofilă și electrofilă la complexele și ale metalelor Ca intermediari ai reacțiilor catalitice, există atât compuși organometalici clasici cu legături M-C, M=C și MC, cât și compuși neclasici în care ligandul organic este coordonat conform 2 , 3 , 4 , 5 și 6 -tip, sau este un element al structurilor cu deficit de electroni - care unește grupările CH 3 și C 6 H 6, carburi neclasice (Rh 6 C(CO) 16, C(AuL) 5 +, C(AuL) 6 2+ etc.). Dintre mecanismele specifice compuşilor -organometalici clasici, remarcăm câteva mecanisme. Astfel, au fost stabilite 5 mecanisme de substituție electrofilă a atomului de metal la legătura M-C. substituție electrofilă cu asistență nucleofilă Adăugare-eliminare AdE(C) Adăugarea la atomul de C în hibridizarea sp 2 AdE(M) Adiție oxidativă la metal Substituția nucleofilă la atomul de carbon în reacțiile de demetalare a compușilor organometalici are loc ca proces redox: Posibilă participare a unui agent oxidant în această etapă Un astfel de agent de oxidare poate fi CuCl2, p-benzochinona, NO3 – și alți compuși. Iată încă două etape elementare caracteristice RMX: hidrogenoliza legăturii M-C și omoliza legăturii M-C O regulă importantă care se aplică tuturor reacțiilor compușilor complecși și organometalici și este asociată cu principiul mișcării minime este regula învelișului de 16-18 electroni a lui Tolman (Secțiunea 2). În mod convențional, reacțiile chimice ale complexelor sunt împărțite în liganzi de schimb, redox, izomerizare și coordonați. Disocierea primară a complexelor în sfera interioară și exterioară determină apariția reacțiilor de schimb ale ionilor din sfera exterioară: X m + mNaY = Y m + mNaX. Componentele sferei interne a complexelor pot participa, de asemenea, la procesele metabolice care implică atât liganzii, cât și agentul de complexare. Pentru a caracteriza reacțiile de substituție ale liganzilor sau ale ionului metalic central, folosiți denumirile și terminologia propuse de K. Ingold pentru reacțiile compușilor organici (Fig. 42), nucleofile. S N și electrofilă S E substituții: Z + Y = z +X S N Z + M"= z + M S E . După mecanismul reacției de substituție, acestea se împart (Fig. 43) în asociative ( S N 1 și SE 1 ) și disociative ( S N 2 și SE 2 ), diferă într-o stare de tranziție cu un număr de coordonare crescut și scăzut. Clasificarea unui mecanism de reacție ca asociativ sau disociativ este o sarcină dificilă, realizabilă experimental, de a identifica un intermediar cu un număr de coordonare redus sau crescut. În acest sens, mecanismul de reacție este adesea judecat pe baza datelor indirecte privind efectul concentrației de reactivi asupra vitezei de reacție, modificări ale structurii geometrice a produsului de reacție etc. Pentru a caracteriza viteza reacțiilor de substituție a ligandului în complexe, laureatul Nobel din 1983 G. Taube (Fig. 44) a propus utilizarea termenilor „labil” și „inert”, în funcție de timpul reacției de substituție a ligandului, mai puțin sau mai mult de 1 minut. . Termenii labili sau inerți sunt caracteristici ale cineticii reacțiilor de substituție a ligandului și nu trebuie confundați cu caracteristicile termodinamice ale stabilității sau instabilității complexelor. Labilitatea sau inerția complecșilor depinde de natura ionului de complexare și a liganzilor. În conformitate cu teoria câmpului ligand: 1.
Complexe octaedrice 3 d metale de tranziție cu distribuție a valenței ( n -1) d electroni pe sigma*(de exemplu ) MO de afânare sunt labile. 4- (t 2g 6 e g 1) + H2O=
3- + CN -. Mai mult, cu cât energia de stabilizare de către câmpul cristalin al complexului este mai mică, cu atât labilitatea acestuia este mai mare. 2.
Complexe octaedrice 3 d metale de tranziție cu sigma liberă* afânare de ex orbitali și distribuția uniformă a valenței ( n -1) d electroni în orbitalii t 2 g (t 2 g 3, t 2 g 6) sunt inerți. [Co III (CN) 6] 3- (t 2 g 6 e g 0) + H 2 O = [Cr III (CN) 6] 3- (t 2 g 3 e g 0) + H 2 O = 3.
Plano-pătrat și octaedric 4 d și 5 d metale de tranziție care nu au electroni per sigma* MO de slăbire sunt inerte. 2+ + H20 = 2+ + H20 = Influența naturii liganzilor asupra vitezei reacțiilor de substituție a liganzilor este considerată în cadrul modelului „influenței reciproce a liganzilor”. Un caz aparte al modelului de influență reciprocă a liganzilor este cel formulat în 1926 de I.I. Conceptul lui Chernyaev de influență trans (Fig. 45) - „labilitatea ligandului în complex depinde de natura ligandului trans-locat” - și propune o serie de influențe trans ale liganzilor: CO, CN -, C 2 H 4 > PR 3, H - > CH 3 -, SC (NH 2) 2 > C 6 H 5 -, NO 2 -, I -, SCN - > Br -, CI - > py , NH3, OH-, H20. Conceptul de influență trans ne-a permis să justificăm regulile generale: 1.
regula lui Peyrone- datorita actiunii amoniacului sau aminelor asupra tetracloroplatinatului ( II ) potasiul se obține întotdeauna diclorodiaminplatină cis-configurație: 2 - + 2NH3 = cis - + 2CI - . Deoarece reacția se desfășoară în două etape și ligandul clorură are o influență trans mare, înlocuirea celui de-al doilea ligand clorură cu amoniac are loc cu formarea de cis-[ Pt (NH3)2Cl2]: 2- + NH3 = - NH3 = cis -. 2.
regula lui Jergensen
- la acțiunea acidului clorhidric asupra clorurii de tetramină de platină ( II ) sau compuși similari se obține configurația trans diclorodi-aminoplatină: [Pt (NH3)4]2+ + 2HCI = trans-[Pt (NH3)2CI2] + 2NH4CI. În conformitate cu seria de influențe trans ale liganzilor, înlocuirea celei de-a doua molecule de amoniac cu un ligand de clorură duce la formarea de trans-[ Pt (NH3)2CI2]. 3.
Reacția cu tiouree a lui Kurnakov
- diferiți produși ai reacției tioureei cu izomerii geometrici ai trans-[ Pt (NH 3 ) 2 Cl 2 ] și cis-[ Pt (NH 3 ) 2 Cl 2 ]: cis - + 4Tio = 2+ + 2CI-+ 2NH3. Natura diferită a produselor de reacție este asociată cu influența trans mare a tioureei. Prima etapă a reacțiilor este înlocuirea liganzilor de clorură de tiouree cu formarea de trans- și cis-[ Pt (NH 3 ) 2 ( Thio ) 2 ] 2+ : trans-[Pt (NH 3) 2 Cl 2 ] + 2 Thio = trans-[ Pt (NH 3) 2 (Tio) 2 ] 2+ cis - + 2Thio = cis - 2+. În cis-[Pt (NH3)2 (Thio ) 2 ] 2+ două molecule de amoniac în poziție trans față de tiouree sunt supuse unei substituții ulterioare, ceea ce duce la formarea 2+ :
cis - 2+ + 2Tio = 2+ + 2NH3. În trans-[Pt (NH3)2 (Thio ) 2 ] 2+ două molecule de amoniac cu influență trans mică sunt situate în poziție trans una față de cealaltă și, prin urmare, nu sunt înlocuite cu tiouree. Modelele de influență trans au fost descoperite de I.I. Chernyaev atunci când studiază reacțiile de substituție a liganzilor în complexe de platină pătrat-planare ( II ). Ulterior, s-a demonstrat că trans-influența liganzilor se manifestă și în complexe de alte metale ( Pt(IV), Pd(II), Co(III), Cr(III), Rh(III), Ir(III). )) și alte structuri geometrice. Adevărat, seria de trans-influență a liganzilor pentru diferite metale sunt oarecum diferite. Trebuie remarcat faptul că influența trans este efect cinetic- cu cât influența trans a unui ligand dat este mai mare, cu atât mai rapid este înlocuit un alt ligand care se află în poziție trans față de acesta. Alături de efectul cinetic al influenței trans, la mijloc XX secolul A.A. Grinberg și Yu.N. Kukushkin a stabilit dependența trans-influenței ligandului L de la ligand situat în poziție cis până la L . Astfel, studiul vitezei de reacție de substituție Cl- amoniac în complexe de platină ( II): [PtCl4] 2- + NH3 = [PtNH3CI3] - + CI - K = 0,42. 10 4 l/mol. Cu [ PtNH 3 Cl 3 ] - + NH 3 = cis-[ Pt (NH 3 ) 2 Cl 2 ] + CI - K = 1,14. 10 4 l/mol. Cu trans-[ Pt (NH 3 ) 2 Cl 2 ] + NH 3 = [ Pt (NH 3 ) 3 Cl ] + + Cl - K = 2,90 . 10 4 l/mol. Cu a arătat că prezența a una sau două molecule de amoniac în poziția cis față de ligand de clorură înlocuit duce la o creștere constantă a vitezei de reacție. Acest efect cinetic se numește influenta cis. În prezent, ambele efecte cinetice ale influenței naturii liganzilor asupra vitezei reacțiilor de substituție a liganzilor (efectul trans și cis) sunt combinate într-un concept general. influența reciprocă a liganzilor.
Fundamentarea teoretică a efectului influenței reciproce a liganzilor este strâns legată de dezvoltarea ideilor despre legăturile chimice în compușii complecși. În anii 30 XX secolul A.A. Greenberg și B.V. Nekrasov a considerat influența trans în cadrul modelului de polarizare: 1.
Efectul trans este tipic pentru complexele al căror ion metalic central este foarte polarizabil. 2.
Activitatea trans a liganzilor este determinată de energia de polarizare reciprocă a ligandului și a ionului metalic. Pentru un ion metalic dat, influența trans a ligandului este determinată de polarizabilitatea acestuia și de distanța față de ionul central. Modelul de polarizare este în concordanță cu datele experimentale pentru complecși cu liganzi anionici simpli, cum ar fi ionii de halogenură. În 1943 A.A. Greenberg a emis ipoteza că activitatea trans a liganzilor este legată de proprietățile lor reducătoare. Schimbarea densității electronilor de la ligandul trans-situat la metal reduce sarcina efectivă a ionului metalic, ceea ce duce la o slăbire a legăturii chimice cu ligandul trans-locat. Dezvoltarea ideilor despre influența trans este asociată cu activitatea trans ridicată a liganzilor bazați pe molecule organice nesaturate precum etilena în [ Pt(C2H4)CI3 ] - . Potrivit lui Chatt și Orgel (Fig. 46), acest lucru se datoreazăpi-interacțiunea dativă a unor astfel de liganzi cu metalul și mecanismul asociativ al reacțiilor de substituție pentru liganzii trans-locați. Coordonarea cu ionul metalic al ligandului atacator Z conduce la formarea unui intermediar bipiramidal trigonal cu cinci coordonate urmat de eliminarea rapidă a ligandului părăsitor X. Formarea unui astfel de intermediar este facilitată depi-interacțiunea ligand dativ-ligand metal Y , care reduce densitatea de electroni a metalului și reduce energia de activare a stării de tranziție cu înlocuirea rapidă ulterioară a ligandului X. Împreună cu p acceptor (C2H4, CN-, CO ...) liganzii care formează o legătură chimică ligand dativ-metal au o mare influență trans șisliganzi donatori: H-, CH3-, C2H5- ... Trans-influența unor astfel de liganzi este determinată de interacțiunea donor-acceptor a ligandului X cu metalul, care scade densitatea electronică a acestuia și slăbește legătura metalului cu ligandul părăsitor Y. Astfel, poziția liganzilor în seria trans-activității este determinată de acțiunea combinată a sigma- donator şi pi-proprietățile liganzilor - sigma- donator şi pi-proprietățile acceptoare ale ligandului își sporesc influența trans, în timp cepi-cei donatori slăbesc. Care dintre aceste componente ale interacțiunii ligand-metal predomină în efectul trans se apreciază pe baza calculelor chimice cuantice ale structurii electronice a stării de tranziție a reacției. Principala reacție de substituție în soluții apoase, schimbul de molecule de apă (22), a fost studiată pentru un număr mare de ioni metalici (Fig. 34). Schimbul de molecule de apă în sfera de coordonare a unui ion metalic cu cea mai mare parte a moleculelor de apă prezente ca solvent are loc foarte rapid pentru majoritatea metalelor și, prin urmare, viteza unei astfel de reacții ar putea fi studiată în principal prin metoda relaxării. Metoda presupune perturbarea echilibrului sistemului, de exemplu printr-o creștere bruscă a temperaturii. În condiții noi (temperatură mai mare), sistemul nu va mai fi în echilibru. Se măsoară apoi rata de echilibru. Dacă puteți modifica temperatura soluției din interior 10 -8 sec, atunci puteți măsura viteza unei reacții care necesită mai mult de o perioadă de timp pentru a se finaliza 10 -8 sec. De asemenea, este posibil să se măsoare viteza de substituție a moleculelor de apă coordonate în diverși ioni metalici cu liganzi SO 2-4, S 2 O 3 2-, EDTA etc. (26). Viteza acestei reacții depinde de concentrația ionului metalic hidratat și nu depinde de concentrația ligandului de intrare, ceea ce face posibilă utilizarea ecuației de ordinul întâi (27) pentru a descrie viteza acestor sisteme. În multe cazuri, viteza de reacție (27) pentru un ion metalic dat nu depinde de natura ligandului de intrare (L), fie că este vorba de molecule de H 2 O sau de SO 4 2-, S 2 O 3 2- sau Ioni EDTA. Această observație, cuplată cu faptul că ecuația vitezei pentru acest proces nu include concentrația ligandului influent, sugerează că aceste reacții au loc printr-un mecanism în care etapa lentă implică ruperea legăturii dintre ionul metalic și apă. Compusul rezultat probabil coordonează rapid liganzii din apropiere. În Sect. 4 din acest capitol s-a afirmat că ionii metalici hidratați mai puternic încărcați, cum ar fi Al 3+ și Sc 3+, schimbă molecule de apă mai lent decât ionii M 2+ și M +; Acest lucru dă motive să presupunem că ruperea legăturilor joacă un rol important în etapa care determină ritmul întregului proces. Concluziile obținute în aceste studii nu sunt concludente, dar dau motive de a crede că procesele S N 1 sunt importante în reacțiile de substituție a ionilor metalici hidratați. Probabil cei mai studiați compuși complecși sunt aminele de cobalt(III). Stabilitatea lor, ușurința de pregătire și reacțiile lente le fac deosebit de potrivite pentru studii cinetice. Deoarece studiile acestor complexe au fost efectuate exclusiv în soluții apoase, ar trebui să luăm în considerare mai întâi reacțiile acestor complexe cu molecule de solvent - apă. S-a constatat că, în general, moleculele de amoniac sau amină coordonate de ionul Co(III) sunt înlocuite atât de lent de molecule de apă, încât se ia în considerare înlocuirea liganzilor alții decât aminele. S-a studiat viteza reacțiilor de tip (28) și s-a constatat că este de ordinul întâi față de complexul de cobalt (X este unul dintre mulți anioni posibili). Deoarece în soluţiile apoase concentraţia de H 2 O este întotdeauna aproximativă 55,5 M, atunci este imposibil de determinat efectul modificării concentrației moleculelor de apă asupra vitezei de reacție. Ecuațiile de viteză (29) și (30) pentru o soluție apoasă nu se pot distinge experimental, deoarece k este pur și simplu egal cu k" = k". Prin urmare, este imposibil de spus din ecuația vitezei de reacție dacă H2O va participa la etapa de determinare a vitezei a procesului. Răspunsul la întrebarea dacă această reacție are loc prin mecanismul S N 2 cu înlocuirea ionului X cu o moleculă de H 2 O sau prin mecanismul S N 1, care implică mai întâi disocierea urmată de adăugarea unei molecule de H 2 O, trebuie poate fi obținută folosind alte date experimentale. Această problemă poate fi rezolvată prin două tipuri de experimente. Viteza de hidroliză (înlocuirea unui ion Cl - pe moleculă de apă) transă- + este de aproximativ 10 3 ori mai mare decât viteza de hidroliză 2+. O creștere a încărcăturii complexului duce la întărirea legăturilor metal-ligand și, în consecință, la inhibarea clivajului acestor legături. De asemenea, ar trebui luate în considerare atracția liganzilor de intrare și facilitarea reacției de substituție. Deoarece a fost găsită o scădere a ratei pe măsură ce sarcina complexului a crescut, în acest caz pare mai probabil un proces disociativ (S N 1). O altă metodă de demonstrare se bazează pe studiul hidrolizei unei serii de complexe similare transă- + . În aceste complexe, molecula de etilendiamină este înlocuită cu diamine similare, în care atomii de hidrogen de la atomul de carbon sunt înlocuiți cu grupări CH3. Complexele care conțin diamine substituite reacţionează mai repede decât complexul de etilendiamină. Înlocuirea atomilor de hidrogen cu grupări CH3 crește volumul ligandului, făcând mai dificil ca atomul de metal să fie atacat de un alt ligand. Aceste obstacole sterice încetinesc reacția prin mecanismul S N 2 Prezența liganzilor voluminoase în apropierea atomului de metal promovează procesul disociativ, deoarece îndepărtarea unuia dintre liganzi reduce acumularea lor la atomul de metal. Creșterea observată a vitezei de hidroliză a complexelor cu liganzi voluminoase este o dovadă bună că reacția se desfășoară conform mecanismului S N 1. Deci, ca urmare a numeroaselor studii ale complexelor de acidoamină Co(II), s-a dovedit că înlocuirea grupărilor de acido cu molecule de apă este un proces disociativ în natură. Legătura atom de cobalt-ligand este extinsă la o anumită valoare critică înainte ca moleculele de apă să înceapă să intre în complex. În complexele cu o sarcină de 2+ și mai mare, ruperea legăturii cobalt-ligand este foarte dificilă, iar intrarea moleculelor de apă începe să joace un rol mai important. S-a constatat că înlocuirea grupării acido (X -) din complexul de cobalt(III) cu o altă grupă decât molecula H2O, (31) trece mai întâi prin înlocuirea acesteia cu o moleculă. solvent - apă, urmat de înlocuirea acestuia cu un nou grup Y (32). Astfel, în multe reacții cu complecși de cobalt(III), viteza de reacție (31) este egală cu viteza de hidroliză (28). Doar ionul hidroxil diferă de ceilalți reactivi prin reactivitatea cu aminele Co(III). Reacționează foarte repede cu complecșii de amine de cobalt(III) (de aproximativ 10 6 ori mai rapid decât apa) în funcție de tipul de reacție hidroliza bazică (33). Această reacţie s-a dovedit a fi de ordinul întâi în ceea ce priveşte ligandul de substituţie OH-(34). Al doilea ordin general al reacției și progresul neobișnuit de rapid al reacției sugerează că ionul OH este un reactiv nucleofil excepțional de eficient pentru complecșii Co(III) și că reacția are loc prin mecanismul S N 2 prin formarea unui intermediar. Totuși, această proprietate a OH - poate fi explicată și printr-un alt mecanism [ecuațiile (35), (36)]. În reacția (35), complexul 2+ se comportă ca un acid (după Brønsted), dând complexul +, care este amido-(conținând)-compus - bază corespunzătoare acidului 2+. Reacția continuă apoi prin mecanismul S N 1 (36) pentru a forma un intermediar cu cinci coordonate, care reacţionează în continuare cu moleculele de solvent pentru a produce produsul final de reacţie (37). Acest mecanism de reacție este în concordanță cu viteza reacției de ordinul doi și corespunde mecanismului S N 1 Deoarece reacția în etapa de determinare a vitezei implică o bază conjugată la complexul original - acidul, acest mecanism primește denumirea S N. 1CB. Determinarea care dintre aceste mecanisme explică cel mai bine observațiile experimentale este foarte dificilă. Cu toate acestea, există dovezi convingătoare care susțin ipoteza S N 1CB. Cele mai bune argumente în favoarea acestui mecanism sunt următoarele: complexele octaedrice Co(III) reacționează în general prin mecanismul disociativ S N 1 și nu există niciun argument convingător de ce ionul OH ar trebui să medieze procesul S N 2 ionul hidroxil este un reactiv nucleofil slab în reacțiile cu Pt(II) și, prin urmare, reactivitatea sa neobișnuită cu Co(III) pare nerezonabilă. Reacțiile cu compuși de cobalt(III) în medii neapoase oferă dovezi excelente pentru formarea intermediarilor cu cinci coordonate furnizați de mecanismul S N 1 SV. Dovada finală este faptul că, în absența legăturilor N - H în complexul Co(III), acesta reacționează lent cu ionii OH -. Acest lucru, desigur, sugerează că proprietățile acido-bazice ale complexului sunt mai importante decât proprietățile nucleofile ale OH pentru viteza de reacție.” Această reacție de hidroliză bazică a complexelor de amine Co(III) ilustrează faptul că datele cinetice poate fi adesea interpretat în mai multe moduri și Pentru a exclude unul sau altul posibil mecanism, este necesar să se efectueze un experiment destul de delicat. În prezent, au fost studiate reacțiile de substituție a unui număr mare de compuși octaedrici. Dacă luăm în considerare mecanismele lor de reacție, cel mai frecvent este procesul disociativ. Acest rezultat nu este neașteptat, deoarece șase liganzi lasă puțin spațiu în jurul atomului central pentru ca alte grupuri să se atașeze de el. Există doar câteva exemple în care a fost demonstrată apariția unui intermediar cu șapte coordonate sau a fost detectată influența unui ligand intermediar. Prin urmare, mecanismul S N 2 nu poate fi complet respins ca o posibilă cale pentru reacțiile de substituție în complexe octaedrice. Etapele elementare ale reacțiilor organice catalizate de acizi, baze, catalizatori nucleofili, complexe metalice, metale solide și compușii acestora în procese eterogene și omogene în fază gazoasă sau lichidă sunt reacții de formare și transformare a diverșilor intermediari organici și organometalici, ca precum și complexe metalice. Compușii organici intermediari includ ioni de carbeniu R+, carboniu RH2+, carbo-anioni R-, cationi anioni și radicali, radicali și biradicali R·, R:, precum și complexe moleculare ale moleculelor donor și acceptor organic (DA), care sunt numite şi de complexe cu transfer de sarcină. În cataliza omogenă și eterogenă prin complecși metalici (cataliza complexă metalică) a reacțiilor organice, intermediarii sunt compuși complecși (de coordonare) cu liganzi organici și anorganici, compuși organometalici cu o legătură M-C, care în majoritatea cazurilor sunt compuși de coordonare. O situație similară apare în cazul chimiei „bidimensionale” pe suprafața catalizatorilor metalici solizi. Să luăm în considerare principalele tipuri de reacții ale complexelor metalice și compușilor organometalici. Reacțiile complexelor metalice pot fi împărțite în trei grupe: a) reacții de transfer de electroni; b) reacţii de substituţie a ligandului; c) reacţii ale liganzilor coordonaţi. Reacții de transfer de electroni Două mecanisme sunt implementate în reacțiile de transfer de electroni - mecanismul sferei exterioare (fără modificări în sferele de coordonare ale donorului și acceptorului) și mecanismul de punte (sfera interioară), ceea ce duce la modificări în sfera de coordonare a metalului. Să luăm în considerare mecanismul sferei exterioare folosind exemplul complexelor octaedrice ale metalelor de tranziție. În cazul reacțiilor simetrice ( G 0 = 0) constantele de viteză variază într-un interval foarte larg de valori - de la 10-12 la 10 5 l mol-1 sec-1, în funcție de configurația electronică a ionului și de gradul de restructurare a acestuia în timpul procesului. În aceste reacții, principiul mișcării minime se manifestă foarte clar - cea mai mică schimbare a învelișului de valență a participanților la reacție. În reacția de transfer de electroni (1) (Co * este un izotop al atomului de Co) (reacție simetrică), Co 2+ (d 7) merge în Co 3+ (d 6). Configurația electronică (shell valent) nu se modifică în timpul acestui transfer 6 electroni la nivelul legăturii triplu degenerate rămân neschimbați (), iar de la nivelul antilegăturii e g electronul de nivelul unu este îndepărtat. Complexul mai puternic Co(NH 3) 6 3+ (mai puternic decât Co(NH 3) 6 2+ ~ 10 30 de ori) este într-o stare de pereche de spin, ca și complexul cu Phen. În acest sens, în procesul de transfer de electroni, învelișul de valență ar trebui reconstruit puternic și, ca urmare, k= 10-9 lmol-1 sec-1. Rata de conversie a Co 2+ în Co 3+, egală cu 50%, se realizează în cazul ligandului Phen în 1 secundă, iar în cazul NH 3 ~ în 30 de ani. Este evident că o etapă cu o astfel de viteză (formal elementară) poate fi exclusă din ansamblul etapelor elementare atunci când se analizează mecanismele de reacție. Magnitudinea G pentru reacția de transfer de electroni în timpul formării unui complex de coliziune, conform teoriei lui Marcus, include două componente și Primul termen este energia de reorganizare a legăturilor M-L în cadrul complexului (lungimea și rezistența legăturii atunci când starea de valență se schimbă). Valoarea include energia de rearanjare a învelișului exterior de solvație în procesul de schimbare a coordonatelor M-L și a încărcăturii complexului. Cu cât este mai mică modificarea mediului electronic și cu cât este mai mică modificarea lungimii M-L, cu atât mai mici sunt liganzii, cu atât este mai mică și, ca urmare, cu atât este mai mare rata de transfer de electroni. Valoarea pentru cazul general poate fi calculată folosind ecuația Marcus Unde. La = 0 . În cazul mecanismului intrasferei, procesul de transfer de electroni este facilitat, deoarece unul dintre liganzii primului complex formează un complex de legătură cu al doilea complex, deplasând unul dintre liganzii din acesta. Constantele de viteză ale unui astfel de proces sunt cu 8 ordine de mărime mai mari decât constantele pentru reducerea Cr(NH 3) 6 3+. În astfel de reacţii, agentul de reducere trebuie să fie un complex labil, iar ligandul din agentul de oxidare trebuie să fie capabil să formeze punţi (Cl-, Br-, I-, N3-, NCS-, bipy). Una dintre cele mai importante etape în cataliza complexului metalic, interacțiunea substratului Y cu complexul, are loc prin trei mecanisme: a) Înlocuirea ligandului cu un solvent. Această etapă este de obicei descrisă ca disocierea complexului Esența procesului în cele mai multe cazuri este înlocuirea ligandului L cu solventul S, care este apoi înlocuit cu ușurință de o moleculă substrat Y. b) Atașarea unui nou ligand la o coordonată liberă cu formarea unui asociat urmată de disocierea ligandului înlocuit c) Substituţie sincronă (tip S N 2) fără formare intermediară În cazul complecșilor Pt(II), viteza de reacție este foarte des descrisă de ecuația cu două căi Unde k SȘi k Y- constantele de viteză ale proceselor care au loc în reacțiile (5) (cu un solvent) și (6) cu ligand Y. De exemplu, Ultima etapă a celei de-a doua rute este suma a trei etape elementare rapide - eliminarea Cl-, adăugarea de Y și eliminarea moleculei de H 2 O. În complexele pătrate plate de metale de tranziție, se observă un efect trans, formulat de I.I Chernyaev - influența LT asupra ratei de substituție a unui ligand care se află în poziție trans față de ligandul LT. Pentru complecșii Pt(II), efectul trans crește în seria de liganzi: H2O~NH3< Cl- ~ Br- < I- ~ NO 2 - ~ C 6 H 5 - < CH 3 - < Prezența efectului trans cinetic și a influenței trans termodinamice explică posibilitatea sintetizării complexelor izomerice inerte de Pt(NH 3) 2 Cl 2: § Reacții de substituție electrofilă (SE) a hidrogenului cu un metal din sfera de coordonare a metalului și procesele lor inverse SH - H2O, ROH, RNH2, RSH, ArH, RCCH. Chiar și moleculele de H2 și CH4 participă la reacții de acest tip § Reacţii de introducere a lui L prin conexiunea M-X În cazul lui X = R (complex organometalic), în legătura M-R se introduc și molecule coordonate cu metal (L - CO, RNC, C 2 H 2, C 2 H 4, N 2, CO 2, O 2 etc. .). Reacția de inserție este rezultatul unui atac intramolecular al nucleofilului X asupra unei molecule coordonate prin - sau -tip. Reacții inverse - - și - reacții de eliminare § Reactii de aditie oxidativa si eliminare reductiva M2 (C2H2) M24+ (C2H2) 4- Aparent, în aceste reacții există întotdeauna o coordonare preliminară a moleculei adăugate, dar aceasta nu poate fi întotdeauna detectată. În acest sens, prezența unui loc liber în sfera de coordonare sau a unui loc asociat cu un solvent, care este ușor înlocuit de un substrat, este un factor important care afectează reactivitatea complecșilor metalici. De exemplu, complecșii bis-alil de Ni sunt buni precursori ai speciilor active catalitic, deoarece datorită eliminării reductive ușoare a bis-alilului, apare un complex cu solventul, așa-numitul. nichel „gol”. Rolul locurilor goale este ilustrat de următorul exemplu: § Reacții de adiție nucleofilă și electrofilă la complecși metalici - și - Ca intermediari ai reacțiilor catalitice, există atât compuși organometalici clasici care au legături M-C, M=C și MC, cât și compuși neclasici în care ligandul organic este coordonat conform tipului 2, 3, 4, 5 și 6 sau este un element cu deficiență de electroni - care unește grupele CH 3 și C 6 H 6, carburi neclasice (Rh 6 C(CO) 16, C(AuL) 5 +, C(AuL) 6 2+ etc.). Dintre mecanismele științifice ale compușilor organometalici clasici, remarcăm câteva mecanisme. Astfel, au fost stabilite 5 mecanisme de substituție electrofilă a atomului de metal la legătura M-C. substituție electrofilă cu asistență nucleofilă AdE Adăugarea-eliminarea AdE(C) Adăugarea la atomul de C în hibridizarea sp 2 AdE(M) Adiție oxidativă la metal Substituția nucleofilă la atomul de carbon în reacțiile de demetalare a compușilor organometalici are loc ca proces de oxidare-reducere: Posibilă participare a unui agent oxidant în această etapă Un astfel de agent de oxidare poate fi CuCl2, p-benzochinona, NO3 - și alți compuși. Iată încă două etape elementare caracteristice RMX: hidrogenoliza legăturii M-C și omoliza legăturii M-C O regulă importantă care se aplică tuturor reacțiilor compușilor complecși și organometalici și este asociată cu principiul mișcării minime este regula învelișului de 16-18 electroni a lui Tolman (Secțiunea 2). Conform conceptelor moderne, pe suprafața metalelor se formează complecși și compuși organometalici similari compușilor din soluții. Pentru chimia de suprafață, participarea mai multor atomi de suprafață la formarea unor astfel de compuși și, desigur, absența particulelor încărcate este esențială. Grupele de suprafață pot fi orice atomi (H, O, N, C), grupuri de atomi (OH, OR, NH, NH 2, CH, CH 2, CH 3, R), molecule coordonate CO, N 2, CO 2, C2H4, C6H6. De exemplu, în timpul adsorbției CO pe o suprafață metalică, s-au găsit următoarele structuri: Molecula C 2 H 4 de pe suprafața metalului formează -complexe cu un centru și punți de etilenă di-conectate M-CH 2 CH 2 -M, i.e. în esenţă cicluri metalice Pe suprafața Rh, de exemplu, în timpul adsorbției etilenei, au loc următoarele procese de conversie a etilenei pe măsură ce temperatura crește: Reacțiile intermediarilor de suprafață includ etapele de adiție oxidativă, eliminare reductivă, inserție, - și -eliminare, hidrogenoliza legăturilor M-C și C-C și alte reacții de tip organometalic, dar fără apariția ionilor liberi. Tabelele prezintă mecanismele și intermediarii transformărilor de suprafață ale hidrocarburilor pe metale. Tabelul 3.1. Reacții catalitice care implică clivajul unei legături C-C. Denumiri: alchil, metalaciclu; Carben, alil; Carabină, vinil. Tabelul 3.2. Reacții catalitice care implică formarea unei legături C-C. Denumiri: vezi tabel. 3.1. Formarea tuturor compușilor organometalici de mai sus pe suprafața metalelor a fost confirmată prin metode fizice. Întrebări pentru autocontrol 1) Cum se manifestă regula celei mai mici modificări în învelișul de valență a unui metal în timpul reacțiilor de transfer de electroni? 2) De ce contribuie posturile vacante de coordonare la interacțiunea eficientă cu substratul? 3) Enumerați principalele tipuri de reacții ale liganzilor coordonați. 4) Prezentați mecanismele de substituție electrofilă în reacțiile compușilor organometalici cu NX. 5) Dați exemple de compuși organometalici de suprafață. 6) Dați exemple de participare a complecșilor de suprafață de carben metalic la transformările hidrocarburilor. Literatură pentru studiu aprofundat 1. Temkin O.N., Kinetics of catalitic reactions in solutions of metal complexs, M., MITHT, 1980, Part III. 2. Collman J., Higedas L., Norton J., Finke R., Organometalic chemistry of consumer metals, M., Mir, 1989, vol. II. 3. Moiseev I.I., -Complexes in the oxidation of olefines, M., Nauka, 1970. 4. Temkin O.N., Shestakov G.K., Treger Yu.A., Acetilena: Chimie. Mecanisme de reacție. Tehnologie. M., Chimie, 1991, 416 p., secțiunea 1. 5. Henrici-Olivet G., Olive S., Coordonare și cataliză, M., Mir, 1980, 421 p. 6. Krylov O.V., Matyshak V.A., Compuși intermediari în cataliză heterogenă, M., Nauka, 1996. 7. Zaera F., Un ghid organometalic pentru chimia motăților de hidrocarburi pe suprafețele metalice de tranziție., Chem. Rev., 1995, 95, 2651 - 2693. 8. Bent B.E., Mimând aspecte ale catalizei heterogene: generarea, izolarea și reacția intermediarilor de suprafață propuși pe cristale individuale în vid, chimie. Rev., 1996, 96, 1361 - 1390. Reacții de substituție, adăugare sau eliminare a liganzilor, în urma cărora sfera de coordonare a metalului se modifică. În sens larg, reacțiile de substituție înseamnă procesele de înlocuire a unor liganzi din sfera de coordonare a unui metal cu alții. Mecanism disociativ (D). În cazul limitativ, un proces în două etape trece printr-un intermediar cu un număr de coordonare mai mic: ML6<->+ L; + Y --» ML5Y Mecanism asociativ (A). Un proces în două etape caracterizat prin formarea unui intermediar cu un număr mare de coordonare: ML6 + Y = ; =ML5Y+L Mecanismul schimbului reciproc (I). Majoritatea reacțiilor de schimb au loc prin acest mecanism. Procesul este într-o etapă și nu este însoțit de formarea unui intermediar. În starea de tranziție, reactivul și grupul de plecare sunt asociate cu centrul de reacție, intră în cea mai apropiată sferă de coordonare a acestuia, iar în timpul reacției un grup este deplasat de altul, are loc un schimb de doi liganzi: ML6 + Y = = ML5Y+L Mecanism intern. Acest mecanism caracterizează procesul de substituție a ligandului la nivel molecular. 2. Caracteristici ale proprietăților lantanidelor (Ln) asociate cu efectul compresiei lantanidelor. Ln 3+ compuși: oxizi, hidroxizi, săruri. Alte stări de oxidare. Exemple de proprietăți reducătoare ale Sm 2+, Eu 2+ și proprietățile oxidante ale Ce 4+, Pr 4+. Scăderea monotonă a razelor atomice și ionice la deplasarea de-a lungul unei serii de elemente 4f se numește compresie lantanidică. eu. Aceasta conduce la faptul că razele atomilor elementelor de tranziție 5d ale grupurilor a patra (hafniu) și a cincea (tantal) care urmează lantanidelor se dovedesc a fi aproape egale cu razele analogilor lor electronici din perioada a cincea: zirconiu. și niobiul, respectiv, iar chimia metalelor grele 4d și 5d are multe în comun. O altă consecință a compresiei f este apropierea razei ionice a ytriului de razele elementelor f grele: disprosium, holmiu și erbiu. Toate elementele pământurilor rare formează oxizi stabili în starea de oxidare +3. Sunt pulberi cristaline refractare care absorb lent dioxidul de carbon și vaporii de apă. Oxizii majorității elementelor sunt obținuți prin calcinarea hidroxizilor, carbonaților, nitraților și oxalaților în aer la o temperatură de 800–1.000 °C. Formează oxizi M2O3 și hidroxizi M(OH)3 Doar hidroxidul de scandiu este amfoter Oxizii și hidroxizii sunt ușor solubili în acizi Sc2O3 + 6HNO3 = 2Sc(NO3)3 + 3H2O Y(OH)3 + 3HCI = YCI3 + 3H2O Numai compușii de scandiu sunt hidrolizați în soluție apoasă Cl3 ⇔ Cl2 + HCl Toate halogenurile sunt cunoscute în starea de oxidare +3. Toate sunt refractare. Fluorurile sunt slab solubile în apă. Y(NO3)3 + 3NaF = YF3↓+ 3NaNO3

Reacții ale liganzilor coordonați
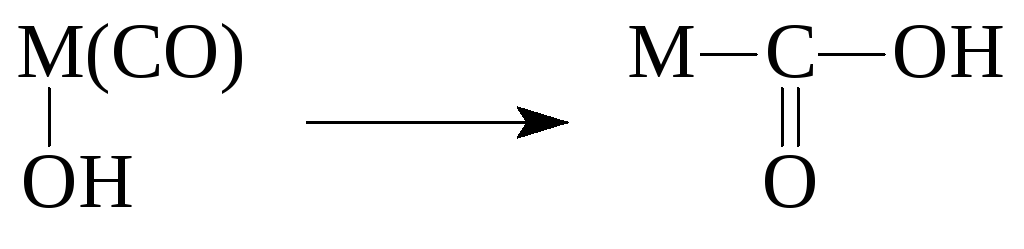









Reacții ale compușilor organometalici



Constanta de viteză de ordinul doi pentru reacție (1) k 1 = 1,1 lmol-1 sec-1. Deoarece Phen (fenantrolina) este un ligand puternic, numărul maxim este de 7 d-electronii sunt perechi (stare spin-paired). În cazul unui ligand slab NH 3 situaţia se schimbă radical. Co(NH3)n2+ (n = 4, 5, 6) este într-o stare de spin-nepereche (spin mare).
< PR 3 ~ AsR 3 ~ H- < олефин ~ CO ~ CN-.